 Overview of OmegaFold and Results
Overview of OmegaFold and ResultsAbstract
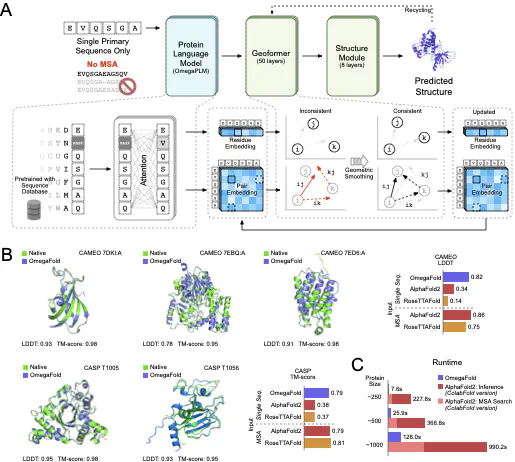
Recent breakthroughs have used deep learning to exploit evolutionary information in multiple sequence alignments (MSAs) to accurately predict protein structures. However, MSAs of homologous proteins are not always available, such as with orphan proteins or fast-evolving proteins like antibodies, and a protein typically folds in a natural setting from its primary amino acid sequence into its three-dimensional structure, suggesting that evolutionary information and MSAs should not be necessary to predict a protein’s folded form. Here, we introduce OmegaFold, the first computational method to successfully predict high-resolution protein structure from a single primary sequence alone. Using a new combination of a protein language model that allows us to make predictions from single sequences and a geometry-inspired transformer model trained on protein structures, OmegaFold outperforms RoseTTAFold and achieves similar prediction accuracy to AlphaFold2 on recently released structures. OmegaFold enables accurate predictions on orphan proteins that do not belong to any functionally characterized protein family and antibodies that tend to have noisy MSAs due to fast evolution. Our study fills a much-encountered gap in structure prediction and brings us a step closer to understanding protein folding in nature.